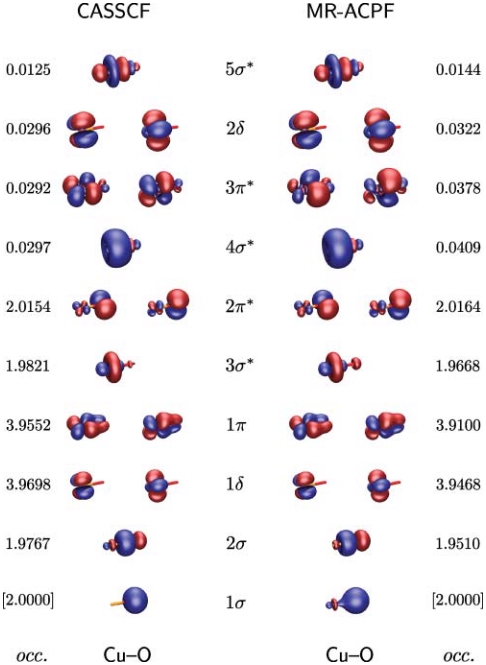
Aerobic Aliphatic Hydroxylation Reactions by Copper Complexes: A Simple Clip‐and‐Cleave Concept
A convenient imine clip‐and‐cleave concept has been developed for the selective hydroxylation of non‐activated C−H bonds of aliphatic aldehydes. Reaction of aldehydes with N,N‐diethyl‐ethylenediamine leads to the corresponding imine that binds copper(I) ions as a bidentate donor ligand. After exposure of a solution of this complex to dioxygen followed by acidic cleavage of the product the corresponding β‐hydroxylated aldehyde forms. This concept was successfully applied to the hydroxylation of trimethylacetaldehyde as well as adamantane and diamantane 1‐carbaldehydes.
Chem. Eur. J. 24, 15543–15549 (2018)
in cooperation with Prof. Schindler (Gießen), Prof. Schreiner (Gießen), and Prof. Fokin (Kiev)

Acid/base triggered interconversion of μ-η2:η2-peroxido and bis(μ-oxido) dicopper intermediates capped by proton-responsive ligands
CuII2(μ-η2:η2-peroxido) and CuIII2(μ-oxido)2cores represent key intermediates in copper/dioxygen chemistry, and they are mechanistically important for biological hydroxylation and oxidation reactions mediated by dinuclear (type III) copper metalloenzymes. While the exact nature of the active species in different enzymes is still under debate, shifting equilibria between Cux/O2 species is increasingly recognized as a means of switching between distinct reactivity patterns of these intermediates. Herein we report comprehensive spectroscopic, crystallographic and computational analysis of a family of synthetic CuII2(μ-η2:η2-peroxido) and CuIII2(μ-oxido)2 dicopper complexes with a bis(oxazoline) (BOX) capping ligand. In particular, we demonstrate that a reversible peroxido/bis(μ-oxido) interconversion of the [Cu2O2] core can be triggered by peripheral (de)protonation events on the ligand backbone. As the copper ions in the enzymes are typically supported by histidine imidazoles that offer a backside N atom amenable to potential (de)protonation, it is well conceivable that the shifting of equilibria between the [Cu2O2] species in response to changes in local pH is biologically relevant.
Chem. Sci. 8, 3031–3037 (2017)
in cooperation with Prof. Meyer (Göttingen)
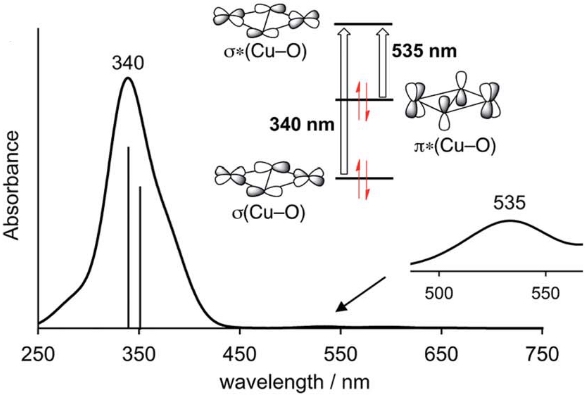
Selective Aromatic Hydroxylation with Dioxygen and Simple Copper Imine Complexes
Introducing OH: A simple copper imine complex system has been developed for selective o-hydroxylation of aromatic aldehydes using dioxygen as the oxidant. By using the ligand N′-benzylidene-N,N-diethylethylenediamine (BDED), salicylaldehyde was prepared in good yields. The underlying reaction mechanism was studied by DFT calculations. The results demonstrate a new facile synthetic way to selectively introduce OH groups into aromatic aldehydes.
Chem. Eur. J. 33, 11735–11744 (2015); Chosen as “Hot Paper”
in cooperation with Prof. Schindler (Gießen), and Prof. Tuczek (Kiel)
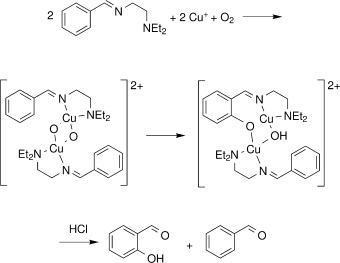
Quantum chemical assessment of the binding energy of CuO+
We present a detailed theoretical investigation on the dissociation energy of CuO+, carried out by means of coupled clustertheory, the multireference averaged coupled pair functional (MR-ACPF) approach, diffusion quantum Monte Carlo (DMC), and density functional theory (DFT). At the respective extrapolated basis set limits, most post-Hartree–Fock approaches agree within a narrow error margin on a De value of 26.0 kcal mol−1[coupled-cluster singles and doubles level augmented by perturbative triples corrections, CCSD(T)], 25.8 kcal mol−1 (CCSDTQ via the high accuracy extrapolated ab initio thermochemistry protocol), and 25.6 kcal mol−1 (DMC), which is encouraging in view of the disaccording data published thus far. The configuration-interaction based MR-ACPF expansion, which includes single and double excitations only, gives a slightly lower value of 24.1 kcal mol−1, indicating that large basis sets and triple excitation patterns are necessary ingredients for a quantitative assessment. Our best estimate for D0 at the CCSD(T) level is 25.3 kcal mol−1, which is somewhat lower than the latest experimental value ( D0 = 31.1 ± 2.8 kcal mol−1; reported by the Armentrout group) [Int. J. Mass Spectrom. 182/183, 99 (1999)]. These highly correlated methods are, however, computationally very demanding, and the results are therefore supplemented with those of more affordable DFT calculations. If used in combination with moderately-sized basis sets, the M05 and M06 hybrid functionals turn out to be promising candidates for studies on much larger systems containing a [CuO]+ core.
J. Chem. Phys.134, 064304 (2011)
in cooperation with Prof. Gauss (Mainz), Dr. Matxain (Donostia), and Prof. Berger (Marburg)