|
|
Research Spotlights
|
|
|
|
|
|
Our central field of research is Computational Chemistry, with a focus on Quantum Chemistry. We apply ab initio or density functional methods to explore reaction mechanisms and to understand the structure and spectroscopy of molecules with unusual electronic and magnetic properties. Our research interests are interdisciplinary and cover areas such as main-group and bioinorganic chemistry, homogenous catalysis, material science, molecular energy carriers, and gas phase chemistry. In recent years our research activities have expanded with a special focus on silicon chemistry and plasma chemistry, fields in which we have started to pursue experimental work as well. Many of our current activities involve close cooperation with experimental groups and industry partners.
|
|
|
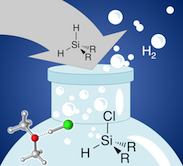
Lewis-Base Catalyzed Selective Chlorination of Monosilanes
HCl/ether at work: A preparatively facile, highly selective synthesis of bifunctional monosilanes R2SiHCl, RSiHCl2 and RSiH2Cl is reported. By chlorination of R2SiH2 and RSiH3 with concentrated HCl/ether solutions the stepwise introduction of Si-Cl bonds is readily controlled by temperature and reaction time for a broad range of substrates. In a combined experimental and computational study, we establish a new mode of Si-H bond activation assisted by Lewis bases such as ethers, amines, phosphines, and chloride ions. Elucidation of the underlying reaction mechanisms shows that alcohol assistance through hydrogen-bond networks is equally efficient and selective. Remarkably, formation of alkoxysilanes or siloxanes is not observed under moderate reaction conditions.
Chem. Eur. J. 2018, doi:10.1002/chem.201803921;
in cooperation with Prof. Auner (Frankfurt)
|
 |
|
|
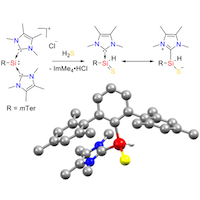
S–H Bond Activation in Hydrogen Sulfide by NHC-Stabilized Silyliumylidene Ions
Reactivity studies of silyliumylidenes remain scarce with only a handful of publications to date. Herein we report the activation of S–H bonds in hydrogen sulfide by mTer-silyliumylidene ion A (mTer = 2,6-Mes2-C6H3, Mes = 2,4,6-Me3-C6H2) to yield an NHC-stabilized thiosilaaldehyde B. The results of NBO and QTAIM analyses suggest a zwitterionic formulation of the product B as the most appropriate. Detailed mechanistic investigations are performed at the M06-L/6-311+G(d,p)(SMD: acetonitrile/benzene)//M06-L/6-311+G(d,p) level of density functional theory. Several pathways for the formation of thiosilaaldehyde B are examined. The energetically preferred route commences with a stepwise addition of H2S to the nucleophilic silicon center. Subsequent NHC dissociation and proton abstraction yields the thiosilaaldehyde in a strongly exergonic reaction. Intermediacy of a chlorosilylene or a thiosilylene is kinetically precluded. With an overall activation barrier of 15 kcal/mol, the resulting mechanistic picture is fully in line with the experimental observation of an instantaneous reaction at sub-zero temperatures.
Inorganics 2018, doi:10.3390/inorganics6020054;
in cooperation with Prof. Inoue (München)
|
 |
|
|
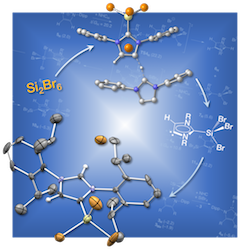
Reactions of Si2Br6 with N‐Heterocyclic Carbenes
A combined experimental and theoretical study on the reaction of Si2Br6 with N‐heterocyclic carbenes is reported. Employment of an imidazole‐2‐ylidene with methyl groups in C4‐ and C5‐position results in the disproportionation of Si2Br6 into the adducts NHC→SiBr2 and NHC→SiBr4. According to expectation, the hydrogenated derivative 1,3‐bis(2,6‐diisopropylphenyl)imidazol‐2‐ylidene forms analogous disproportionation products of Si2Br6 at low temperatures, whereas reaction at higher temperatures furnishes the 4‐SiBr3‐substituted NHC. The underlying formation mechanism explored by means of density functional theory calculations features an abnormal carbene intermediate.
Z. Anorg. Allg. Chem. 2018, 17, 982–988;
in cooperation with Prof. Auner (Frankfurt)
|
 |
|
|
Thermal Synthesis of Perchlorinated Oligosilanes: A Fresh Look at an Old Reaction
Old reaction, new tricks: A complex mixture of small oligosilanes is produced by quenching the thermal reaction of SiCl4 and elemental silicon. A combined theoretical and experimental reinvestigation of the long-known (SiCl2)x synthesis sheds new light on the reaction product and its reactivity. The Supporting Information provides, among other things, a comprehensive compilation of computed 29Si NMR chemical shifts for cyclic and acyclic perchlorosilanes, most of which have not been studied experimentally yet.
Chem. Eur. J. 2017, 23, 12399–12405
in cooperation with Prof. Auner (Frankfurt)
|
 |
|
|
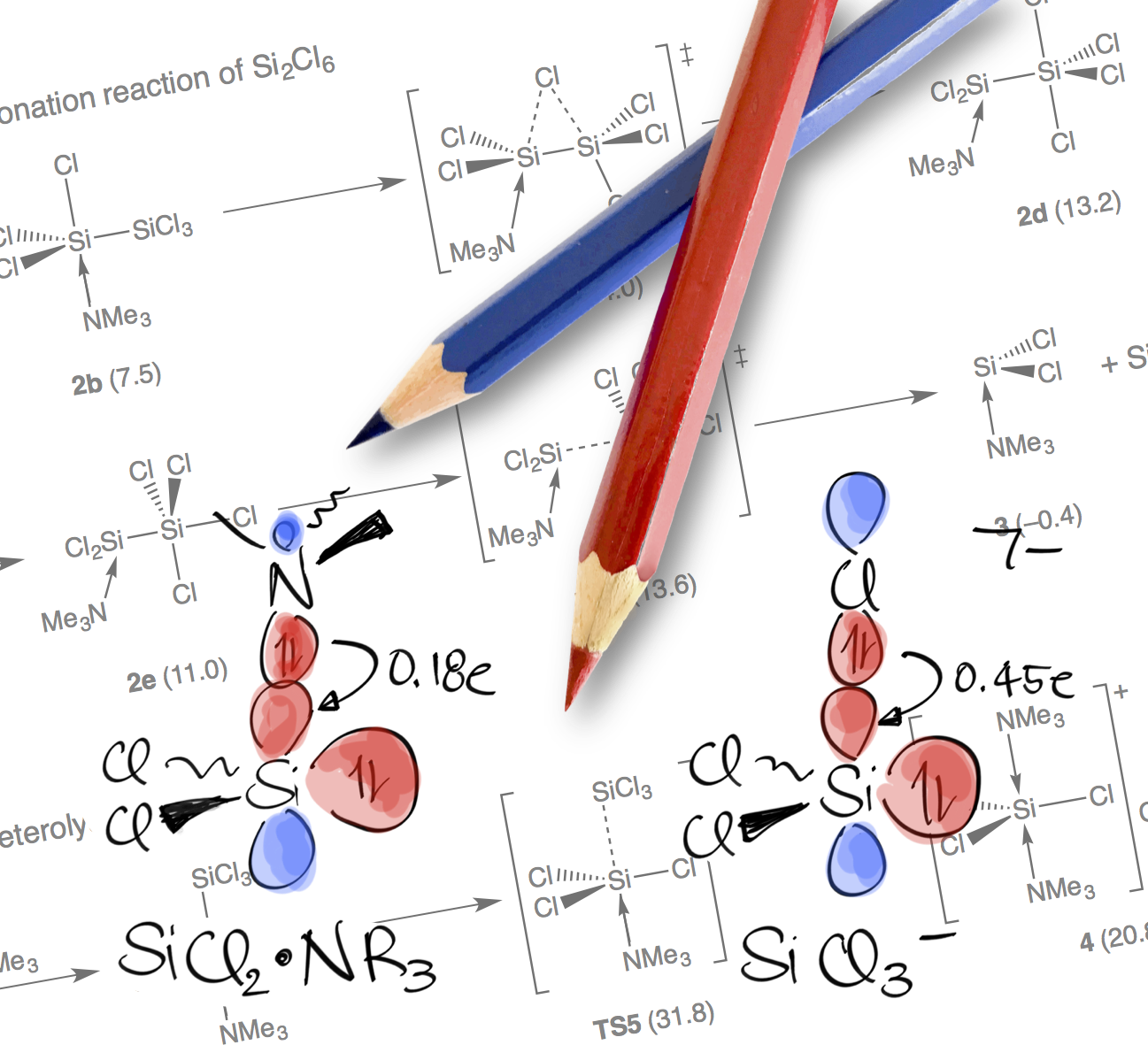
Unraveling the Amine-Induced Disproportionation Reaction of Perchlorinated Silanes—A DFT Study
A neo twist: A DFT study on the amine-induced disproportionation reaction of Si2Cl6 to neo-Si5Cl12 discloses a stepwise rather than a concerted silylene insertion mechanism, which was generally accepted for over half a century. The resulting picture appears generalizable to the related chloride-induced chemistry recently explored.
Chem. Eur. J. 2016, 22, 14328 –14335; Chosen as "Hot Paper"
|
 |
|
|
|
|
|
|
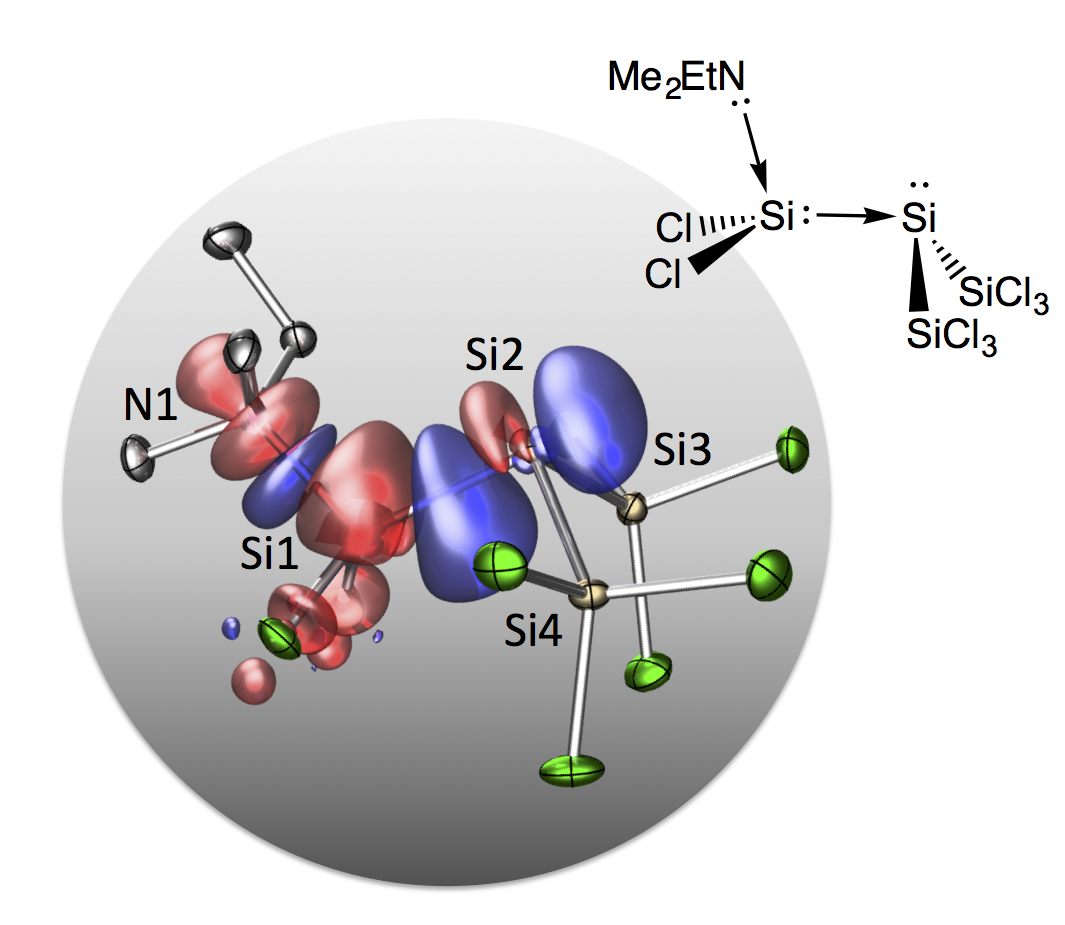
A Disilene Base Adduct with a Dative Si–Si Single Bond
Give and take: A perchlorodisilene amine adduct with an unusually bent structure was isolated by freeze-quench crystallization. The quantum-chemical bond analysis discloses dative N1![[RIGHTWARDS ARROW]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/002192.gif) Si1 and Si1Si2 bonds leading to a push–pull stabilization of the central SiCl2 group. Si1 and Si1Si2 bonds leading to a push–pull stabilization of the central SiCl2 group.
Angew. Chem. Int. Ed. 2016, 55,1782 –1786; Angew. Chem. 2016, 128, 1814 –1818
in cooperation with Prof. Schneider (Göttingen), Dr. Linser (MPI Göttingen), and Prof. Auner (Frankfurt)
|
 |
|
|
|
|
|
|
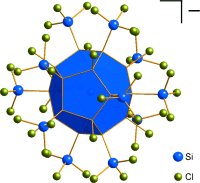
One-Step Synthesis of a [20]Silafullerane with an Endohedral Chloride Ion
As simple as this: A stable, crystalline [20]-silafullerane forms in preparatively useful yields through wet-chemical self-assembly from Si2Cl6 and chloride ions in the presence of an amine. Each silicon dodecahedron contains an endohedral chloride ion that imparts a net negative charge. Eight chloro substituents and twelve trichlorosilyl groups are attached to the surface of each cluster in a strictly regioregular arrangement.
Angew. Chem. Int. Ed. 2015, 54, 5429 –5433; Highlighted as "Very Important Paper"
in cooperation with Prof. Wagner (Frankfurt)
|
|
|
|
|
|
|
|
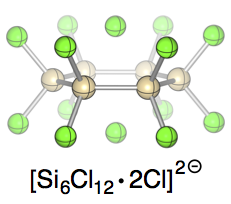
Chloride-induced aufbau of perchlorinated cyclohexasilanes from Si2Cl6 – A mechanistic scenario
As simple as this: Chloride addition to Si2Cl6 provides efficient synthetic access to perchlorocyclohexasilane dichloride adducts. The reaction mechanism has been elucidated in a combined theoretical and experimental study: chloride-induced SiCl3− liberation from Si2Cl6 triggers the formation of higher silanide anions, which dimerize in a head-to-tail fashion to the cyclic final products.
Chem. Eur. J. 2014, 20, 9234 –9239; Conference Issue: 17th ISOS, Berlin
in cooperation with Prof. Wagner (Frankfurt)
|
|
|
|
|
|
|
|
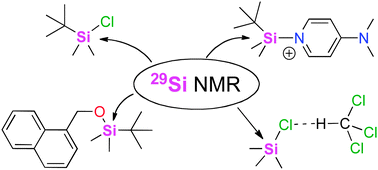
The calculation of 29Si NMR chemical shifts of tetracoordinated silicon compounds in the gas phase and in solution
Aiming at the identification of an efficient computational protocol for the accurate NMR assessment of organosilanes in low-polarity organic solvents, 29Si NMR chemical shifts of a selected set of such species relevant in organic synthesis have been calculated relative to tetramethylsilane (TMS, 1) using selected density functional and perturbation theory methods. Satisfactory results are obtained when using triple zeta quality basis sets such as IGLO-III. Solvent effects impact the calculated results through both, changes in substrate geometry as well as changes in the actual shieldings. Spin–orbit (SO) corrections are required for systems carrying more than one chlorine atom directly bonded to silicon. Best overall results are obtained using gas phase geometries optimized at MPW1K/6-31+G(d) level in combination with shielding calculations performed at MPW1K/IGLO-III level in the presence of the PCM continuum solvation model.
Phys. Chem. Chem. Phys. 2014, 16, 16642 –16650
in cooperation with Prof. Zipse (München)
|
 |
|
|
|
|
|
|
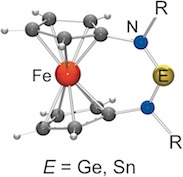
Redox‐Active N‐Heterocyclic Germylenes and Stannylenes with a Ferrocene‐1,1′‐diyl Backbone
Be positive, tetrylene! N‐heterocyclic germylenes and stannylenes with a ferrocenylene backbone are described, which are the first examples of redox‐functionalised N‐heterocyclic terylenes (see figure). Consistent with the strongly localised nature of the HOMO, the oxidation process is ferrocene based. The ferrocenium‐type nature of the resulting cation does not compromise the fundamental tetrylene character of these species, thus opening the door to a reversible umpolung of their electronic profile by redox switching.
Chem. Eur. J. 2017, 23, 1187-1199
in cooperation with Prof. Siemeling (Kassel)
|
 |
|
|
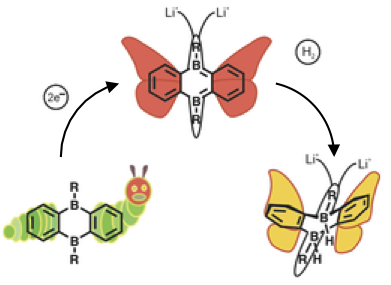
Reversible Dihydrogen Activation by Reduced Aryl Boranes as Main-Group Ambiphiles
Borane metamorphosis: The injection of two electrons transformed the ditopic Lewis acid diboraanthracene into a main-group ambiphile (MGA) that activated H2 much as a transition metal would. The H2 molecule underwent selective addition across the two boron atoms, and the resulting hydridoborate could be used as a hydride-transfer reagent.
Angew. Chem. Int.Ed. 2016, DOI: 10.1002/anie.201608324
in cooperation with Prof. Wagner (Frankfurt)
|
 |
|
|
|
|
|
|
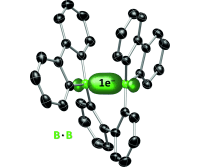
Confirmed by X-ray crystallography: The B•B one-electron σ bond
One is enough: The first structurally characterized radical anion containing a B⋅B one-electron σ bond shows a significantly shorter B⋅⋅⋅B distance than the uncharged starting material, while the boron centers largely maintain their local planarity.
Angew. Chem. Int. Ed. 2014, 53, 4832 –4835; Highlighted as "Very Important Paper"
in cooperation with Prof. Wagner (Frankfurt)
|
 |
|
|
|
|
|
|
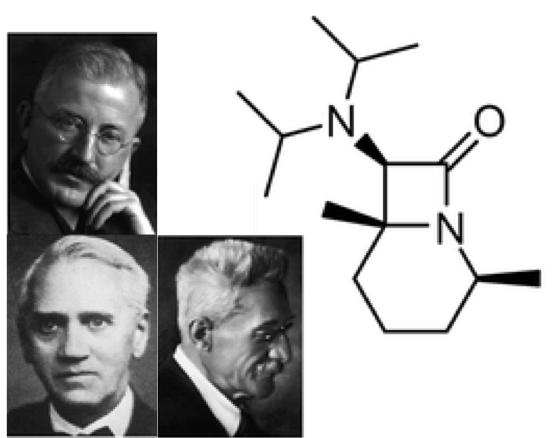
Diastereoselective synthesis of a bicyclic β-lactam with penicillin G-like spectrum of activity by carbonylation of an acyclic diaminocarbene
Diisopropylamino-cis-2,6-dimethylpiperidinocarbene reacts regio- and diastereoselectively with CO to afford a bicyclic β-lactam with 100% atom efficiency, whose spectrum of activity resembles that of penicillin G or amoxicillin.
Chem. Commun., 2014, 50, 2341 -2343
in cooperation with Prof. Siemeling (Kassel), Prof. Bandow (Bochum), and Prof. Schneider (Bonn)
|
|
|
|
|
|
|
|
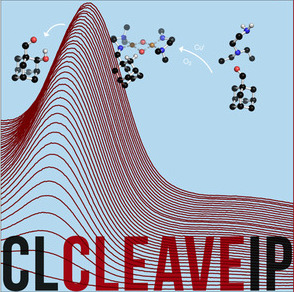
Aerobic Aliphatic Hydroxylation Reactions by Copper Complexes: A Simple Clip‐and‐Cleave Concept
A convenient imine clip‐and‐cleave concept: : has been developed for the selective hydroxylation of non‐activated C−H bonds of aliphatic aldehydes. Reaction of aldehydes with N,N‐diethyl‐ethylenediamine leads to the corresponding imine that binds copper(I) ions as a bidentate donor ligand. After exposure of a solution of this complex to dioxygen followed by acidic cleavage of the product the corresponding β‐hydroxylated aldehyde forms. This concept was successfully applied to the hydroxylation of trimethylacetaldehyde as well as adamantane and diamantane 1‐carbaldehydes.
Chem. Eur. J. 2018, https://doi.org/10.1002/chem.201802607
in cooperation with Prof. Schindler (Gießen), Prof. Schreiner (Gießen) und Prof. Fokin (Kiev)
|
 |
|
|
|
|
|
|
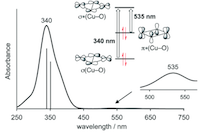
Acid/base triggered interconversion of μ-η2:η2-peroxido and bis(μ-oxido) dicopper intermediates capped by proton-responsive ligands
CuII2(μ-η2:η2-peroxido) and CuIII2(μ-oxido)2cores represent key intermediates in copper/dioxygen chemistry, and they are mechanistically important for biological hydroxylation and oxidation reactions mediated by dinuclear (type III) copper metalloenzymes. While the exact nature of the active species in different enzymes is still under debate, shifting equilibria between Cux/O2 species is increasingly recognized as a means of switching between distinct reactivity patterns of these intermediates. Herein we report comprehensive spectroscopic, crystallographic and computational analysis of a family of synthetic CuII2(μ-η2:η2-peroxido) and CuIII2(μ-oxido)2 dicopper complexes with a bis(oxazoline) (BOX) capping ligand. In particular, we demonstrate that a reversible peroxido/bis(μ-oxido) interconversion of the [Cu2O2] core can be triggered by peripheral (de)protonation events on the ligand backbone. As the copper ions in the enzymes are typically supported by histidine imidazoles that offer a backside N atom amenable to potential (de)protonation, it is well conceivable that the shifting of equilibria between the [Cu2O2] species in response to changes in local pH is biologically relevant.
Chem. Sci. 2017, 8, 3031-3037;
in cooperation with Prof. Meyer (Göttingen)
|
 |
|
|
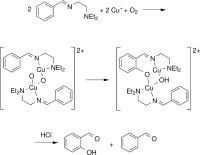
Selective Aromatic Hydroxylation with Dioxygen and Simple Copper Imine Complexes
Introducing OH: A simple copper imine complex system has been developed for selective o-hydroxylation of aromatic aldehydes using dioxygen as the oxidant. By using the ligand N′-benzylidene-N,N-diethylethylenediamine (BDED), salicylaldehyde was prepared in good yields. The underlying reaction mechanism was studied by DFT calculations. The results demonstrate a new facile synthetic way to selectively introduce OH groups into aromatic aldehydes.
Chem. Eur. J. 2015, 33, 11735–11744; Chosen as "Hot Paper"
in cooperation with Prof. Schindler (Gießen), and Prof. Tuczek (Kiel)
|
 |
|
|
|
|
|
|
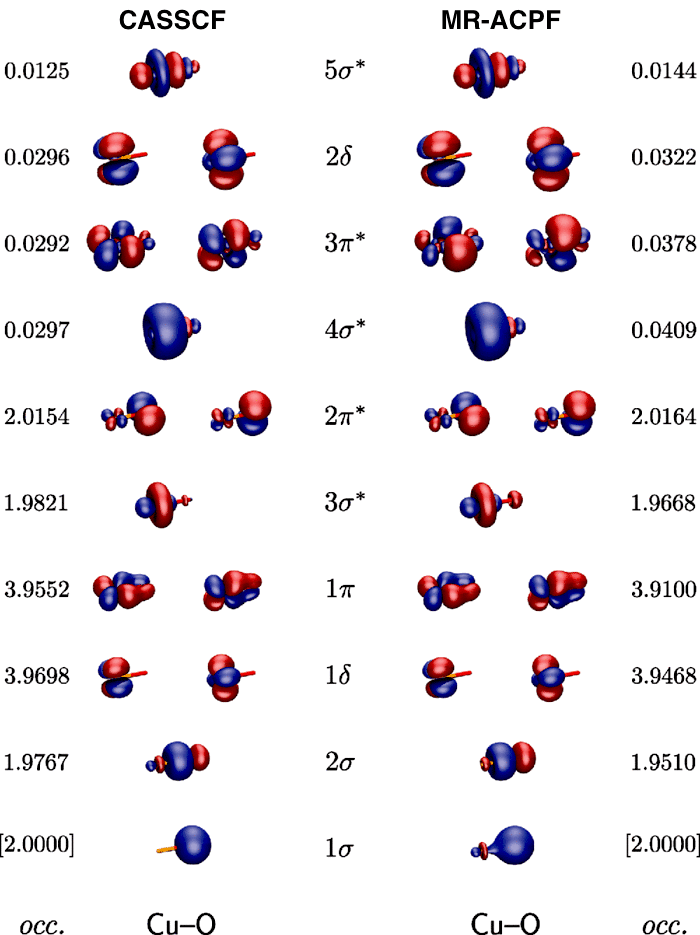
Quantum chemical assessment of the binding energy of CuO+
We present a detailed theoretical investigation on the dissociation energy of CuO+, carried out by means of coupled clustertheory, the multireference averaged coupled pair functional (MR-ACPF) approach, diffusion quantum Monte Carlo (DMC), and density functional theory(DFT). At the respective extrapolated basis set limits, most post-Hartree–Fock approaches agree within a narrow error margin on a De value of 26.0 kcal mol−1[coupled-cluster singles and doubles level augmented by perturbative triples corrections, CCSD(T)], 25.8 kcal mol−1 (CCSDTQ via the high accuracy extrapolated ab initio thermochemistry protocol), and 25.6 kcal mol−1 (DMC), which is encouraging in view of the disaccording data published thus far. The configuration-interaction based MR-ACPF expansion, which includes single and double excitations only, gives a slightly lower value of 24.1 kcal mol−1, indicating that large basis sets and triple excitation patterns are necessary ingredients for a quantitative assessment. Our best estimate for D0 at the CCSD(T) level is 25.3 kcal mol−1, which is somewhat lower than the latest experimental value ( D0 = 31.1 ± 2.8 kcal mol−1; reported by the Armentrout group) [Int. J. Mass Spectrom. 182/183, 99 (1999)]. These highly correlated methods are, however, computationally very demanding, and the results are therefore supplemented with those of more affordable DFT calculations. If used in combination with moderately-sized basis sets, the M05 and M06 hybrid functionals turn out to be promising candidates for studies on much larger systems containing a [CuO]+ core.
J. Chem. Phys. 2011, 134, 064304
in cooperation with Prof. Gauss (Mainz), Dr. Matxain (Donostia), and Prof. Berger (Marburg)
|
 |
|
|
|
|
|
|
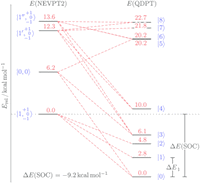
An iridium(III/IV/V) redox series featuring a terminal imido complex with triplet ground state
The iridium(III/IV/V) imido redox series [Ir(NtBu){N(CHCHPtBu2)2}]0/+/2+ was synthesized and examined spectroscopically, magnetically, crystallographically and computationally. The monocationic iridium(IV) imide exhibits an electronic doublet ground state with considerable ‘imidyl’ character as a result of covalent Ir–NtBu bonding. Reduction gives the neutral imide [Ir(NtBu){N(CHCHPtBu2)2}] as the first example of an iridium complex with a triplet ground state. Its reactivity with respect to nitrene transfer to selected electrophiles (CO2) and nucleophiles (PMe3), respectively, is reported.
Figure. Computed state-energy diagram of [Ir(NtBu)(PNP)] . Relative energies of the lowest non-relativistic (left) and spin–orbit states (right) in red (cm−1) and corresponding |S, MS〉 labels in blue.
Chem. Sci. 2018, 9, 4325-4332
in cooperation with Prof. Schneider (Göttingen), Prof. de Bruin (Amsterdam) und Prof. van Slageren (Stuttgart)
|
 |
|
|
|
|
|
|
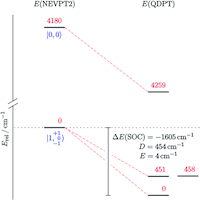
A square-planar osmium(II) complex
Reduction of the pincer complex [OsIIICl2(PNP)] (PNP = N(CHCHPtBu2)2) affords the isolation and full characterization of an osmium(II) complex with square-planar coordination geometry, i.e. [OsIICl(PNP)]. Spectroscopic, structural and magnetic data in combination with multireference computations indicate strong temperature independent paramagnetism, which arises from an energetically well separated ground state that mixes with excited states through spin–orbit coupling.
Figure. State-energy diagram for [Os[OsIICl(PNP)] based on NEVPT2/SA-CASSCF(16,10) computations. Non-relativistic energies of the lowest four states are shown with their corresponding |S,MS〉 label (left). The lowest nine spin–orbit states (right) are complemented with selected contributions (weights >10%) of the corresponding spin-free states (dashed red lines).
Chem. Commun. 2017, 53, 5511-5514
in cooperation with Prof. Schneider (Göttingen)
|
|
|
|
|
|
|
|
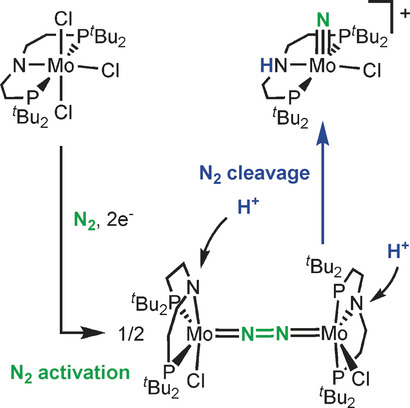
Dinitrogen Splitting Coupled to Protonation
Acid splits: Protonation of an N2‐bridged dimolybdenum complex in the pincer periphery results in splitting into MoV nitrides. This proton‐coupled metal‐to‐ligand charge transfer reaction provides a mechanism to control the thermochemistry and kinetics of N−N bond cleavage with proton‐responsive ligands.
Angew. Chem. Int. Ed. 2017, 56, 5872-5876
in cooperation with Prof. Schneider (Göttingen)
|
 |
|
|
|
|
|
|
|
|
|
|
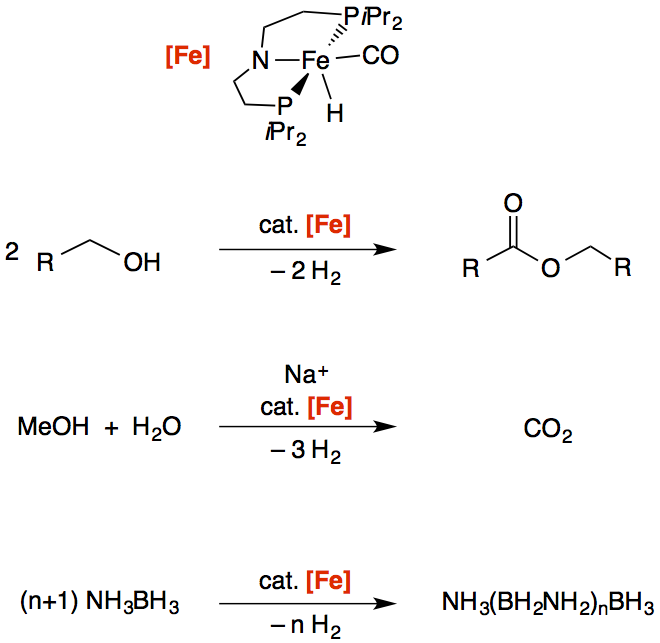
Fe-PNP Mediated Dehydrogenation Catalysis
Chemical hydrogen storage materials: The pincer-supported iron catalyst [Fe] was successfully employed in highly-efficient dehydrogenation reactions of different substrates. The development of iron-based homogeneous catalysts for these applications (i.e. dehydrogenation of potential chemical hydrogen storage materials) is a major advancement in this field, since conventional approaches used precious or heavy metals exclusively.
ACS Catal. 2015, 5, 7214 –7217
ACS Catal. 2015, 5, 2404 –2415; Highlighted in ACS Catal. 2015, 5, 5584 –5585
ACS Catal. 2014, 4, 3994 –4003
in cooperation with Prof. Schneider (Göttingen), Prof. Schmedt a. d. Günne (Siegen), Prof. Bernskoetter (Brown University), Prof. Hazari (Yale University), and Prof. Jones (Rochester)
|
 |
|
|
|
|
|
|
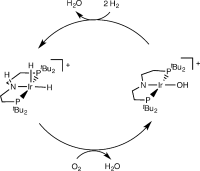
Oxygen Reduction with a Bifunctional Iridium Dihydride Complex
A mononuclear mechanism: The oxygen-reduction reaction (ORR) with an iridium dihydride results in formation of an unusual square-planar iridium(III) hydroxide and water. The dihydride is regenerated with H2 in a quasi-catalytic synthetic cycle. Experimental and computational studies are in agreement with a four-electron ORR mechanism at a single metal site.
Angew. Chem. Int. Ed. 2015, 54,15271 –15275; Angew. Chem. 2015, 127, 15486 –15490
in cooperation with Prof. Schneider (Göttingen)
|
 |
|
|
|
|
|
|
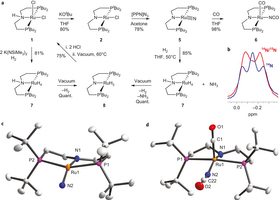
Ammonia formation by metal–ligand cooperative hydrogenolysis of a nitrido ligand
Bioinspired hydrogenation of N2 to ammonia at ambient conditions by stepwise nitrogen protonation/reduction with metal complexes in solution has experienced remarkable progress. In contrast, the highly desirable direct hydrogenation with H2 remains difficult. In analogy to the heterogeneously catalysed Haber–Bosch process, such a reaction is conceivable via metal-centred N2 splitting and unprecedented hydrogenolysis of the nitrido ligands to ammonia. We report the synthesis of a ruthenium(IV) nitrido complex. The high nucleophilicity of the nitrido ligand is demonstrated by unusual N–C coupling with π-acidic CO. Furthermore, the terminal nitrido ligand undergoes facile hydrogenolysis with H2 at ambient conditions to produce ammonia in high yield. Kinetic and quantum chemical examinations of this reaction suggest cooperative behaviour of a phosphorus–nitrogen–phosphorus pincer ligand in rate-determining heterolytic hydrogen splitting.
Nature Chemistry 2011, 3, 532 –537; Highlighted in Nature Chemistry 2011, 3, 502 –504 and Chemistry World, May 25, 2011
in cooperation with Prof. Schneider (Göttingen), and Dr. Herdtweck (München)
|
 |
|
|
|
|
Datenschutz nach Artikel 13 EU DSGVO
|
